


Background
The primary goal of most phylogenetic analyses in BEAST is to infer the posterior distribution of trees and associated model parameters using Markov chain Monte Carlo (MCMC) sampling. In this tutorial, we will learn how to analyze the output of an MCMC analysis in BEAST2 using Tracer. This program allows us to easily visualize BEAST’s output and summarize results. As we will see, we can also use Tracer to troubleshoot some of the most common MCMC problems encountered in BEAST.
While BEAST’s MCMC algorithm is fairly well optimised for phylogenetic inference, problems can arise, especially as the complexity of our data and models increase. An MCMC run may not converge on a stationary target distribution. More commonly, a run might converge but mix poorly - meaning that our samples from the posterior are highly autocorrelated and therefore not independent. In these cases, it is often necessary to tune the performance of the MCMC algorithm.
In this tutorial, we will consider a relatively simple example where we would like to infer a phylogeny and evolutionary parameters from a small alignment of sequences. Our job will be to work together to increase the efficiency of the MCMC so we can make BEAST purr…
Programs used in this Exercise
BEAST2 - Bayesian Evolutionary Analysis Sampling Trees 2
BEAST2 (http://www.beast2.org) is a free software package for Bayesian evolutionary analysis of molecular sequences using MCMC and strictly oriented toward inference using rooted, time-measured phylogenetic trees. This tutorial is written for BEAST v2.7.x (Bouckaert et al., 2014; Bouckaert et al., 2019).
BEAUti2 - Bayesian Evolutionary Analysis Utility
BEAUti2 is a graphical user interface tool for generating BEAST2 XML configuration files.
Both BEAST2 and BEAUti2 are Java programs, which means that the exact same code runs on all platforms. For us it simply means that the interface will be the same on all platforms. The screenshots used in this tutorial are taken on a Mac OS X computer; however, both programs will have the same layout and functionality on both Windows and Linux. BEAUti2 is provided as a part of the BEAST2 package so you do not need to install it separately.
Tracer
Tracer (http://beast.community/tracer) is used to summarise the posterior estimates of the various parameters sampled by the Markov Chain. This program can be used for visual inspection and to assess convergence. It helps to quickly view median estimates and 95% highest posterior density intervals of the parameters, and calculates the effective sample sizes (ESS) of parameters. It can also be used to investigate potential parameter correlations. We will be using Tracer v1.7.x.
Practical: Getting BEAST to purr
The data
To get started, I have generated a XML file that we can run our phylogenetic analysis in BEAST.
Download the first BEAST2 input file
tutorial_run1.xml
The XML contains an alignment of 36 randomly simulated DNA sequences.
Inspecting the XML file in BEAUti
While we can open the XML file in any standard text editor, BEAUTi offers an easy way to inspect the different elements of the analysis:
Open BEAUti and load in the
tutorial_run1.xmlfile by navigating to File > Load.
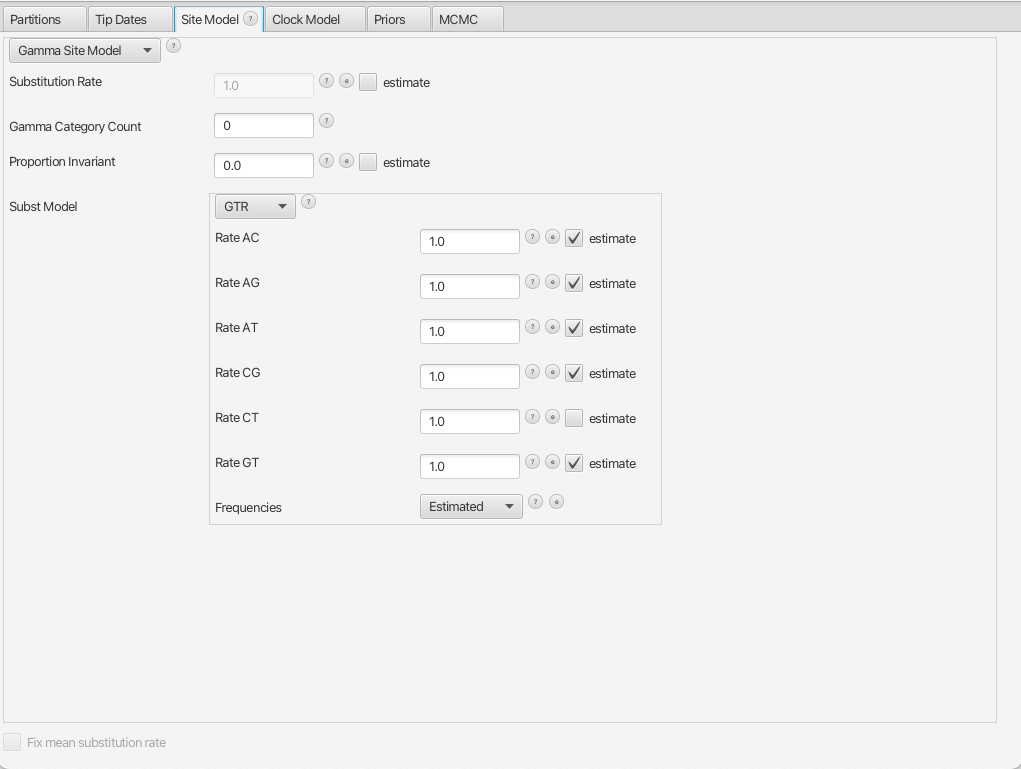
By navigating between the different tabs at the top of the application window, we can inspect the data and each element of the analysis. For example, in the Site Model panel, we can see that we are fitting a GTR substitution model with no gamma rate heterogeneity (Figure 1).
Running the XML in BEAST
We are now ready to run our first analysis in BEAST.
Open BEAST2 and choose
tutorial_run1.xmlas the Input file. Then click Run.
Since we are only running 200,000 iterations, the MCMC should finish running in under 30 seconds.
Visualizing BEAST’s output in Tracer
Open Tracer and navigate to File > Import Trace File, then open
tutorial_run1.logor simply drag and drop the file into the Tracer window.
The results of your run will look similar, but not necessarily identical, to my results shown in Figure 2.
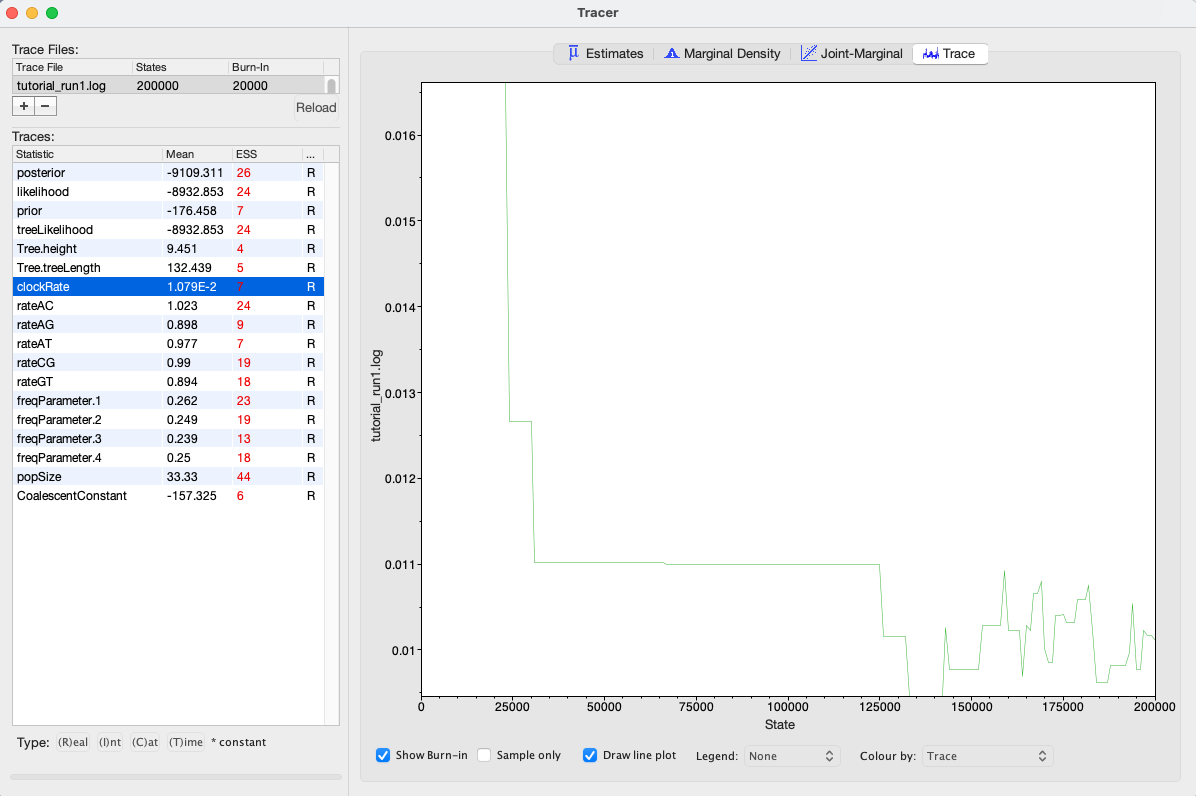
Tracer allows us to quickly visualize BEAST’s MCMC output in order to check performance and see our parameter estimates. On the left there is a panel where all model parameters are listed along with their mean posterior estimates and Effective Sample Size (ESS). Recall that the ESS tells us how many pseudo-independent samples we have from the posterior, so the higher the better. Here, we can see that the ESS is low for all parameters, indicating that we do not yet have a good estimate of the posterior distribution.
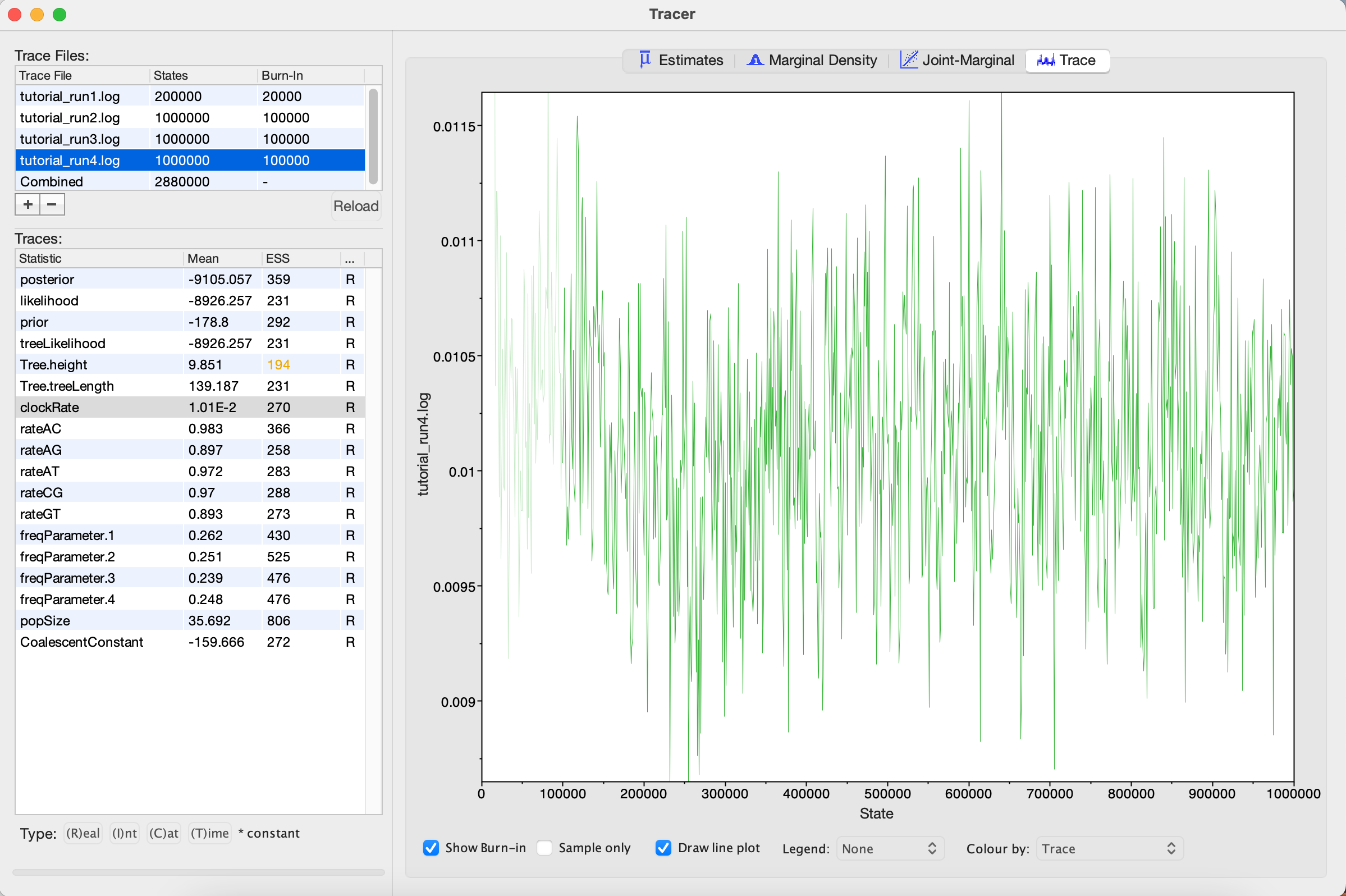
By selecting a parameter in the left panel and then clicking on the Trace tab, we can see how the MCMC explored parameter space (Figure 3). For the clockRate parameter for instance, we see that the chain quickly converged to a value of about 0.01 (the true value used to simulate the sequence data), but mixing was poor, hence the low ESS.
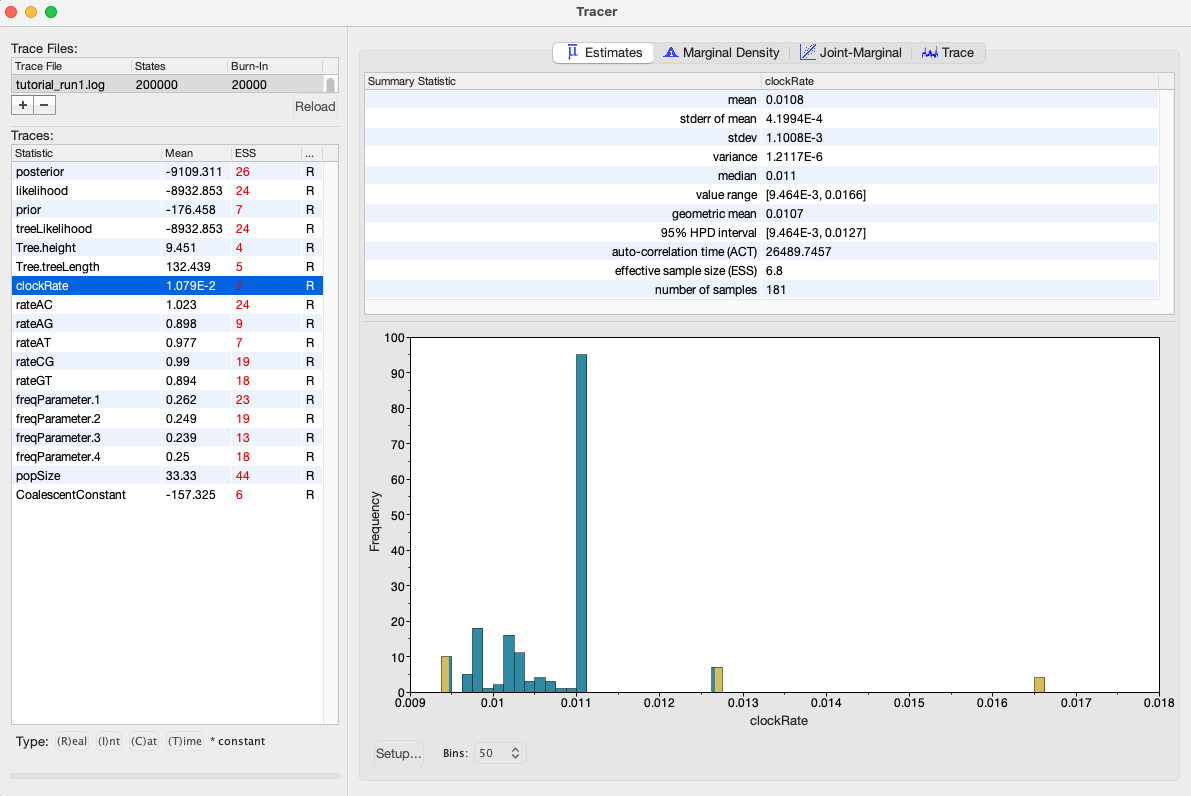
We can also see our posterior estimates for each parameter by clicking on the Estimates tab while highlighting the desired parameter in the left panel. This provides us with various summary statistics and a frequency histogram representing our estimate of the posterior distribution constructed from our MCMC samples. For the clockRate parameter, we can see that our estimate of the posterior is extremely rough, again because we have so few uncorrelated samples from the posterior (Figure 4).
Run 2: Increasing the chain length
By checking the ESS, trace plots and parameter estimates, we got the picture that none of our parameters in Run 1 mixed well. In this case, the simplest thing to try is to rerun the MCMC for more iterations.
Load
tutorial_run1.xmlback into BEAUti using File > Load. Navigate to the MCMC panel and increase the chain length to 1 million. When done, navigate File > Save As and save astutorial_run2.xml.
Note that we didn’t need to change the file names fo the tracelog or treelog, since they are by default set to $(filebase).log and $(filebase)-$(tree).trees. When running the analysis $(filebase) will be replaced by the name of the XML file and $(tree) by the name of tree in the analysis (in this case the name of the alignment).
Run the
tutorial_run2.xmlfile in BEAST2 as we did before. When done, opentutorial_run2.login Tracer.
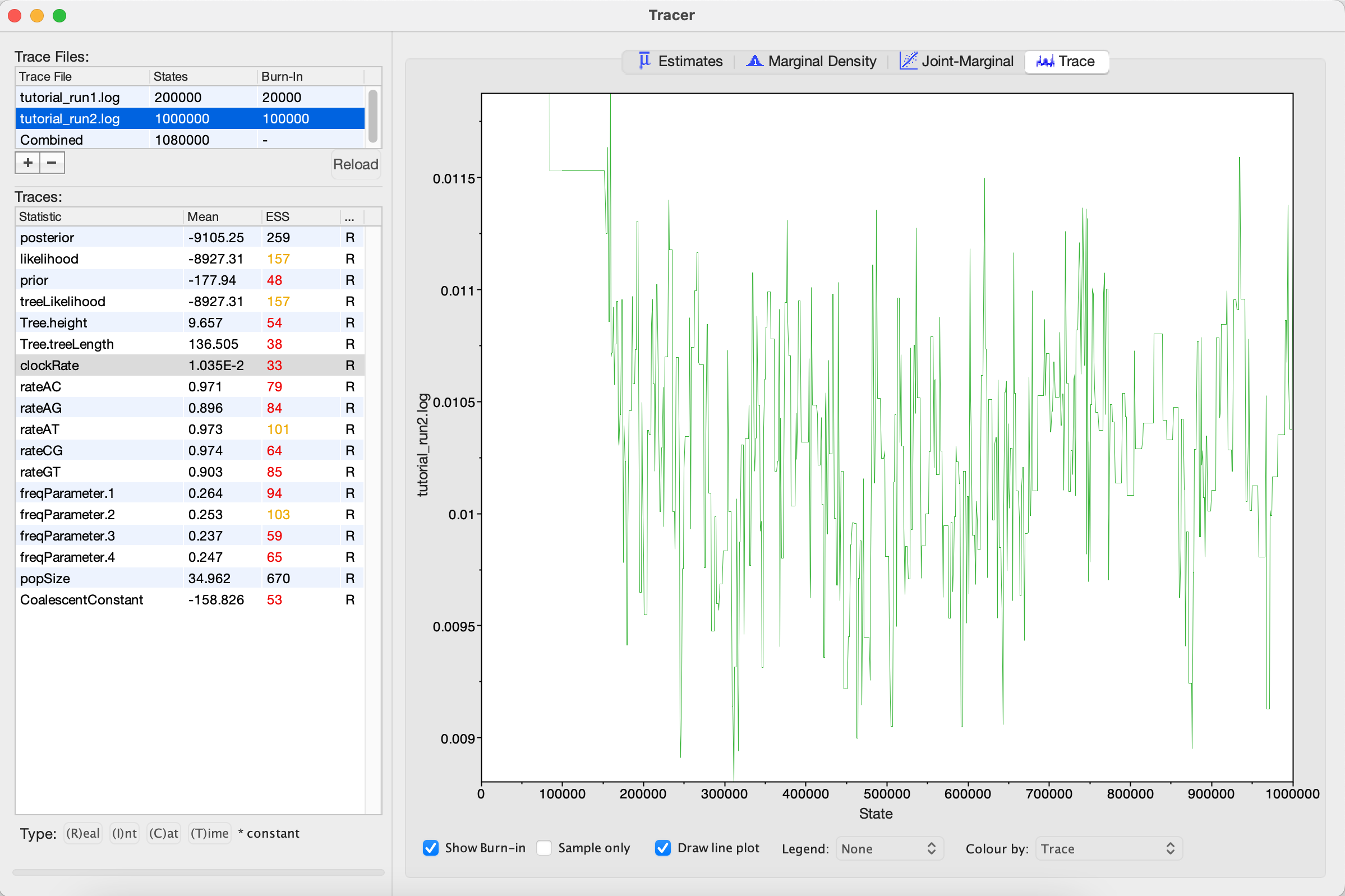
Looking at the MCMC output in Tracer, we see that increasing the chain length did help (Figure 5). The ESS values are higher and the traces look better, but still not great. In the next section, we will continue to focus on the clockRate parameter because it still has a low ESS and appears to mix especially poorly.
Run 3: Changing the clockRate operators
If one parameter in particular is not converging or mixing well, we can try to tweak that parameter’s operator(s). Remember that BEAST’s operators control what new parameter values are proposed at each MCMC iteration and how these proposals are made (i.e. the proposal distribution). Since the clockRate parameter was not mixing well in Run 2, we will try increasing the frequency at which new clockRates are proposed.
Load
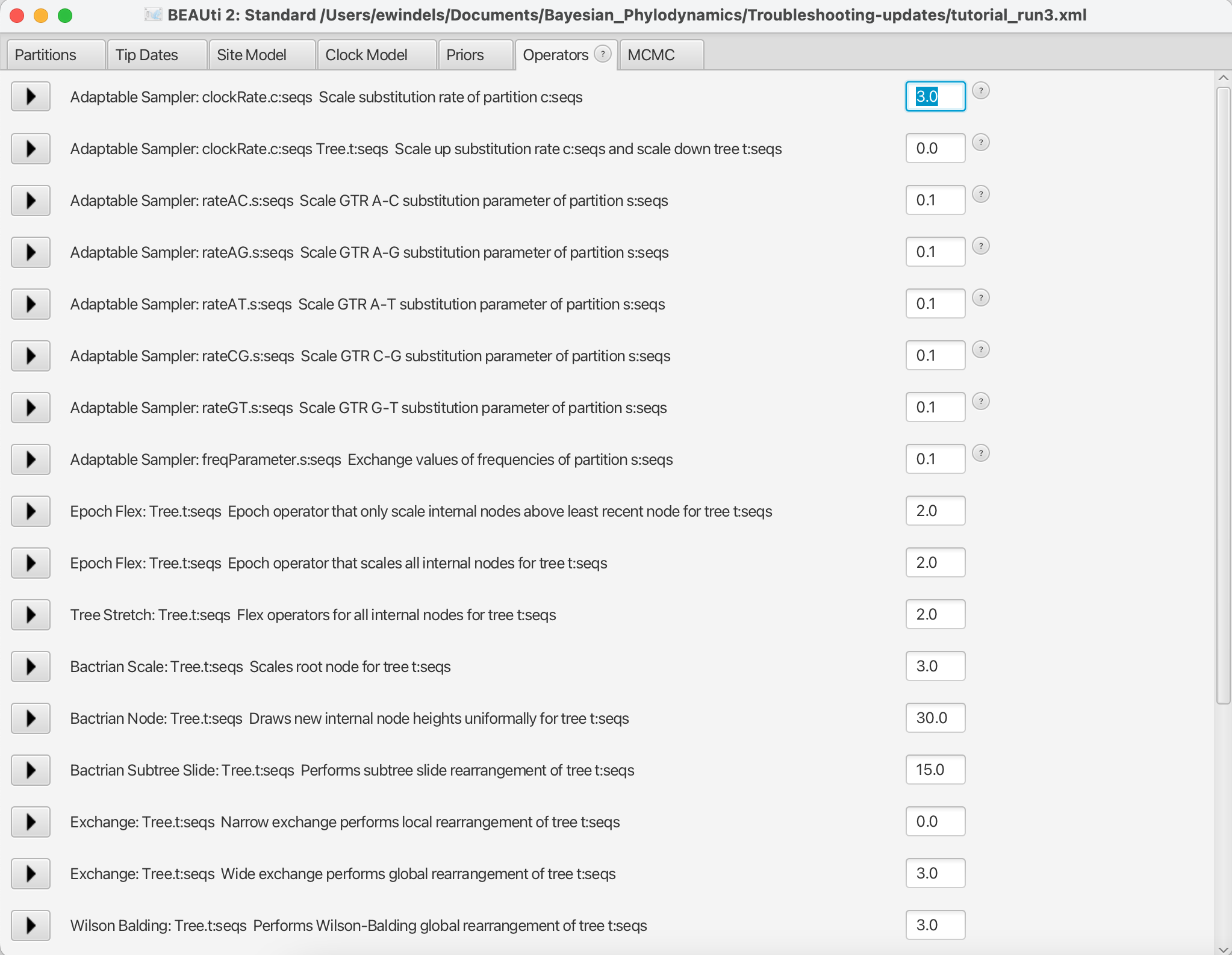
tutorial_run2.xmlback into BEAUti and select View > Show Operators panel. This will bring up a new panel showing all the operators in use (Figure 6). In the box to the right ofAdaptable Sampler: clockRate.c:seqs Scale substitution rate of partition c:seqs, change the value from 0.05 to 3.0. When done, save astutorial_run3.xml.
So, what just happened? We increased the weight of the scale operator on the clockRate, which moves the parameter value up or down, so that new clockRates will be proposed more often in the MCMC. Going from a weight of 0.05 to 3.0 means that new proposals for that parameter will be made sixty times as often, but the frequency at which a given operator is called depends on the weights given to other operators. So if there are parameters with very high ESS values, we may want to reallocate weight on their operators to operators on less well mixing parameters.
Run
tutorial_run3.xmlin BEAST2 and then opentutorial_run3.login Tracer.
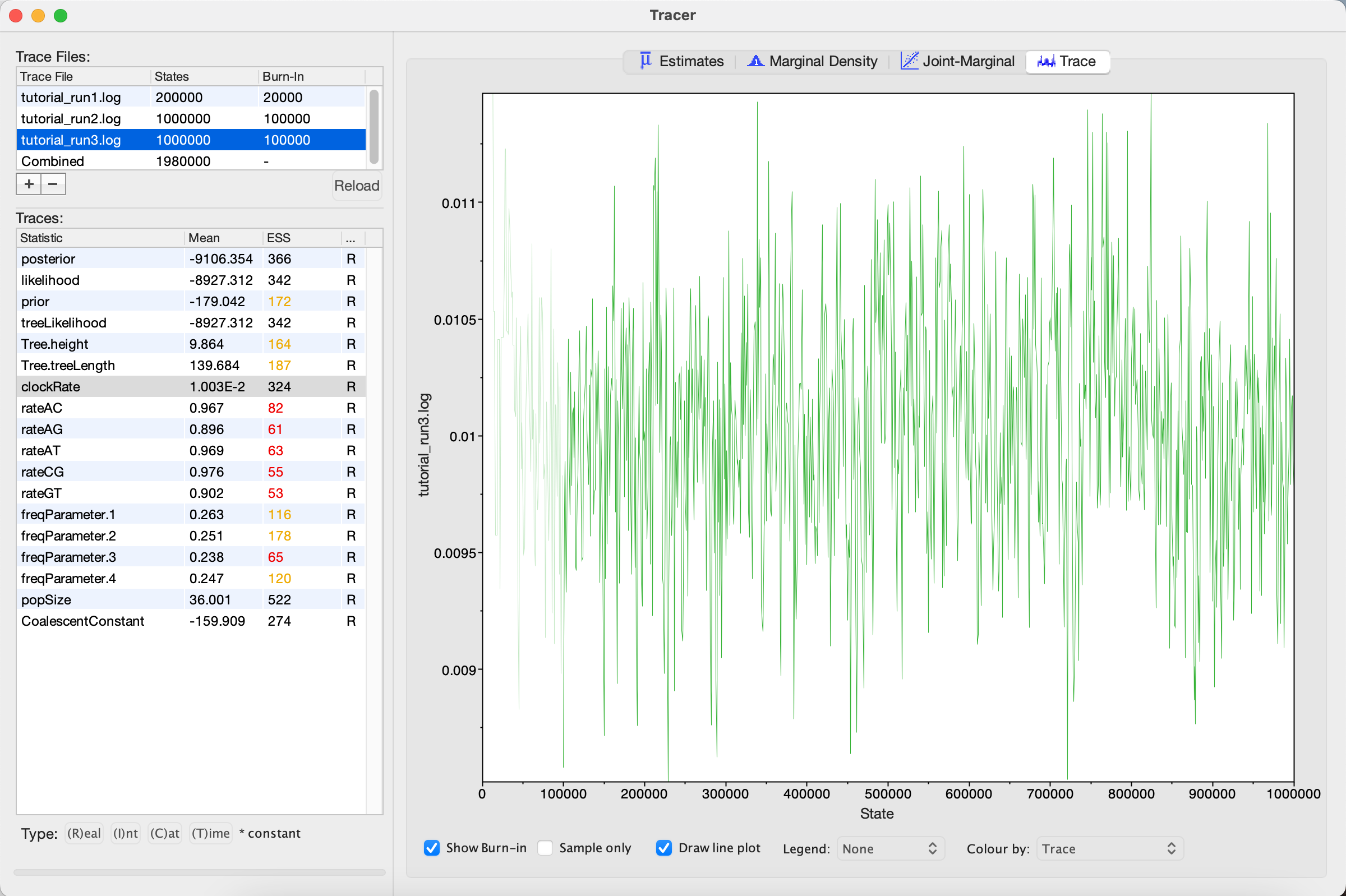
We can see that optimizing the operator dramatically improves mixing for the clockRate (Figure 7).
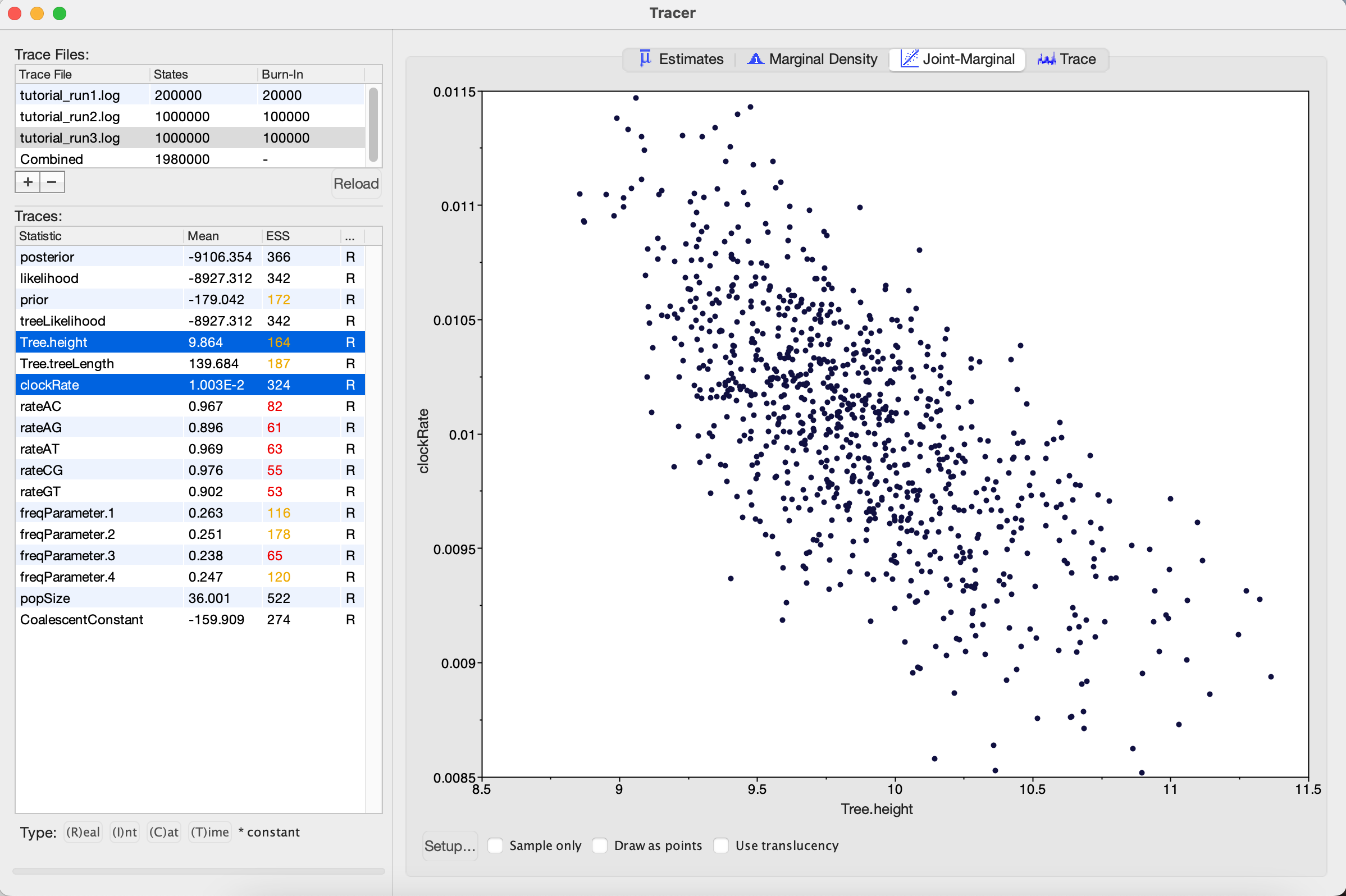
One thing to keep in mind is that BEAST is using MCMC to explore a multidimensional parameter space, and poor mixing in one dimension can be caused by poor mixing in another dimension. This often arises because two parameters are highly correlated. We can identify these correlations in Tracer by visualizing the joint distribution of a pair of parameters together. To do this, select one parameter in the left panel and then, while holding the command key (Mac) or control key (Windows), select another. Then click on the Joint-Marginal tab at the top and uncheck the Sample only box at the bottom. Looking at the pairwise joint distribution for Tree.height and clockRate, we see that these two parameters are highly negatively correlated (Figure 8). We therefore may want to add an operator that updates these parameters together to more efficiently explore their parameter space.
Run 4: Adding an UpDown operator
The easiest way to improve MCMC performance when two parameters are highly negatively correlated is to add an UpDown operator. This operator scales one parameter up while scaling the other parameter down. If two parameters are highly positively correlated we can also use this operator to scale both parameters in the same direction, up or down.
Load
tutorial_run3.xmlback into BEAUti and select View > Show Operators panel as before. In the box to the right ofAdaptable Sampler: clockRate.c:seqs Tree.t:seqs Scale up substitution rate c:seqs and scale down tree t:seqs, change the weight on the Bactrian UpDown operator from 0.0 to 3.0. When done, save astutorial_run4.xml.
We just added an UpDown operator on the clockRate and the Tree.height parameters. The fact that these two parameters are highly negatively correlated makes perfect sense. An increase in the clockRate means that less time is needed for substitutions to accumulate along branches; meaning branches can be shorter and yet still explain the same amount of accumulated evolutionary change in the sequence data. If all branches in the tree become shorter, then the total Tree.height will also decrease. Thus, it makes sense to include an UpDown operator on clockRate and Tree.height. In fact, by default BEAUTi includes this operator. However, I disabled it in the original XML file by setting the weight on this operator to zero for the purpose of illustration.
Run
tutorial_run4.xmlin BEAST2 and then opentutorial_run4.login Tracer.
Looking at the MCMC output in Tracer, we see that all parameters are starting to mix well with relatively high ESS values. Personally, I would probably want to run one final MCMC for several million iterations just to be on the safe side, but this can easily be done by adding more iterations to the chain as we did for Run 2. Alternatively, multiple different MCMC runs can be combined using the program LogCombiner that comes packaged with BEAST. This may be better than running one single long analysis, as it allows us to be sure independent runs are converging on posterior parameter values and increases our confidence that we are sampling from the equilibrium distribution.
Further things to keep in mind
- The number of MCMC iterations needed to achieve a reasonable posterior sample in this tutorial was quite small. With larger alignments, much longer chains may be needed.
- In this tutorial we only considered MCMC performance with respect to exploring parameter space, but we also need to consider tree space. One simple diagnostic for checking convergence and mixing in tree space is to look at the trace plot for the tree likelihood. Poor mixing in the tree likelihood can indicate problems exploring tree space.
- It is always a good idea to check your posterior estimates against sampling from the prior.
Practical: Using a starting tree
By default, BEAST2 generates a random starting tree for you at the beginning of the analysis. While this is helpful, the random starting tree can be quite far from the phylogenies with high posterior values. Using a good initial tree, for instance a phylogeny which was generated during a previous analysis of the dataset, can be a good way to make the burn-in phase shorter and accelerate the convergence of our analysis. Here we use a primates dataset with only 12 sequences as an example, but providing a starting tree will generally be more effective for larger phylogenies and more complex analyses.
The Data
As in the previous section, we have already generated an XML file containing all the details of our analysis.
Download the first BEAST2 input file
primates_run.xml.
Inspecting the XML file in BEAUti
While we can open the XML file in any standard text editor, BEAUTi offers an easy way to inspect the different elements of the analysis:
Open BEAUti and load in the
primates_run.xmlfile by navigating to File > Load.
By default, the initial value for the tree does not appear in BEAUti, so our first step is to make this initial tree visible.
In BEAUti, navigate to View > Show Starting tree panel. A new tab appears in the interface, which should look like Figure 10.
Setting an initial tree
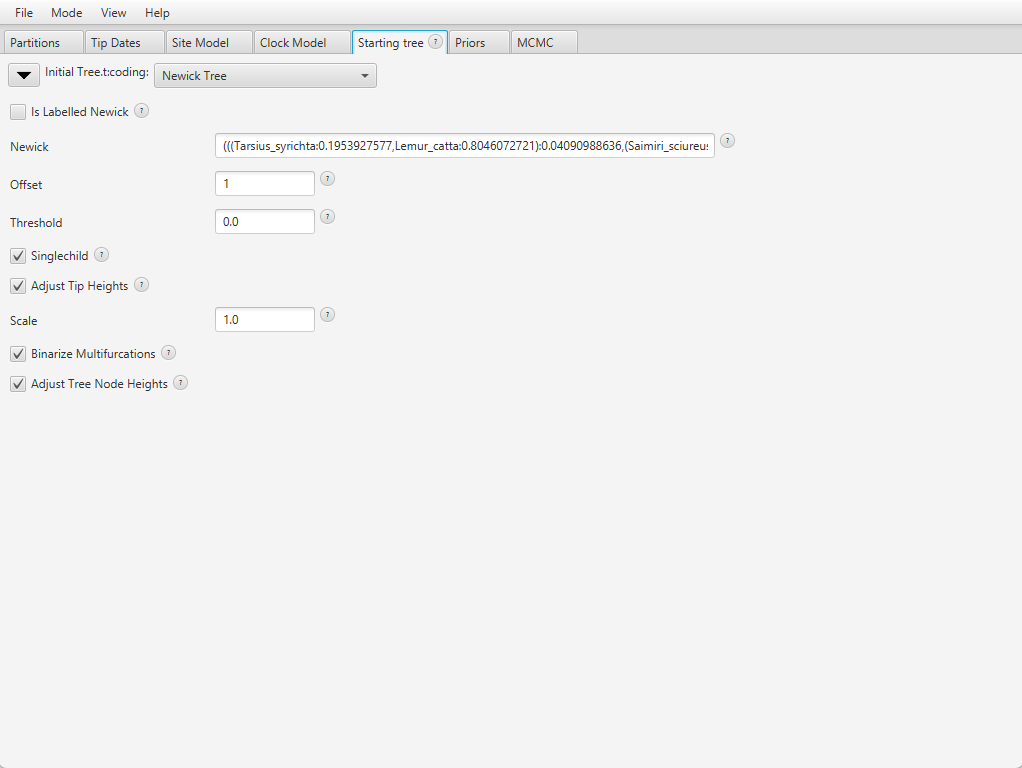
Our first step is to select a newick tree as our starting tree, which will allow us to input directly our initial tree.
Click on the dropdown menu indicating Random Tree and select the option Newick Tree.
We now need to provide our initial tree in Newick format.
Download the first initial tree file
primates_tree.tre. Copy and paste the tree from the file into the Newick input field.
Note that there are several important options in this menu, which you need to consider carefully as they can modify your input tree:
- Adjust Tip Heights: if tip dates are not provided for your sequences and this option is checked, the ages of all the tips in your tree will be adjusted to 0 (i.e. the present). If you have a tree with heterochronous samples or fossils and you did not provide the tip ages (in the Tip ages panel), you need to uncheck this option, otherwise your tree will be changed to an ultrametric phylogeny.
- Binarize Multifurcations: this option will remove any polytomies present in your initial tree and resolve them as a series of random bifurcations. Since most tree models implemented in BEAST2 cannot handle polytomies, this option is set to true by default.
- Adjust Tree Node Heights: this option will adjust branch lengths which are negative or zero to small positive values. Sampled ancestors are generally encoded as tips at the end of branches with length zero, so you need to uncheck this option if you want to provide an initial tree with sampled ancestors.
Our analysis only contains extant species, and our tree has no polytomies so we will leave all options to their default. The final setup looks like Figure 11.
Running with an initial tree
We can now run our updated analysis.
Save the analysis as
primates_run_newick.xml. Open BEAST2 and chooseprimates_run_newick.xmlas the Input file. Then click Run.
We can load the log files primates_run.log and primates_run_newick.log (found in the Output section of the tutorial) into Tracer to compare both runs. On this small example, the ESS of the run with an initial tree are slightly higher on most parameters than with the random tree, but both runs have converged and we do not see a strong difference in performance from adding an initial tree. However, initial trees can be very helpful for larger and more complex inferences to converge.
Common issues with initial trees
A frequent issue with using an initial tree is that BEAST2 expects a dated tree, i.e. a tree with branch lengths in units of time, whereas initial trees are often generated using a maximum likelihood software such as IQ-TREE, which will output a tree with branch lengths in units of number of substitutions. This is exactly the case in our example, and if we plot the primates tree provided above we will notice this very easily. For instance, all our samples come from the present, but our primates initial tree is not ultrametric.
This was not an issue in our earlier inference because BEAST2 was able to correct the tip ages (with the Adjust Tip Heights option) and we had no other time calibration points. However, in a real analysis we will likely have several calibrations to help calibrate our molecular clock. Let us check what happens when we add a root calibration at the root of the tree.
Download the second BEAST2 input file
primates_run_newick_root.xml. Open BEAUti and load in theprimates_run_newick_root.xmlfile by navigating to File > Load. Check the added root calibration in the Priors panel.
We can see here that we have added a distribution on the root node in order to infer a dated tree. Now we can run this analysis.
Open BEAST2 and choose
primates_run_newick_root.xmlas the Input file. Then click Run.
Our analysis does not start, and by looking at the error message in Figure 12 we can see that our root calibration has probability -Infinity. This is because the root calibration that we have set has an offset of 20 My, meaning that the root age has to be above that value. On the other hand, the initial tree we have provided has a root age of 0.887, which is completely incompatible with the calibration.

We can solve this issue by scaling our initial tree so that the root age is increased. Since our root age has to be greater than 20, we need a scaling factor of at least 20/0.887 = 22.55 to have a tree which is compatible with the calibration.
Open BEAUti and load in the
primates_run_newick_root.xmlfile by navigating to File > Load. As before, navigate to View > Show Starting tree panel. Set the Scale to 25.0.
We can now test that our updated analysis runs correctly.
Save the analysis as
primates_run_newick_root_scaled.xml. Open BEAST2 and chooseprimates_run_newick_root_scaled.xmlas the Input file. Then click Run.
Unfortunately, the Scale parameter is applied to the whole tree, and in analyses with many different calibration points it is not always possible to find a scale factor which is compatible with all calibrations, especially if some calibrations have both a minimum and a maximum age. For instance, let us see what happens if we add a second calibration point to our analysis.
Download the third BEAST2 input file
primates_run_newick_2calib.xml. Open BEAUti and load in theprimates_run_newick_2calib.xmlfile by navigating to File > Load. Check the two calibrations in the Priors panel.
We can see two calibrations in our analysis, one at the root of the tree and one on the human-chimpanzee clade. Now we can run this analysis.
Open BEAST2 and choose
primates_run_newick_2calib.xmlas the Input file. Then click Run.
Again our analysis does not start, and by looking at the error message in Figure 13 we can see that our second calibration human_pan has probability -Infinity, which indicates that it is incompatible with the initial tree. In this situation, we would need to either use a dated tree as our starting value for the inference, or manually adjust the tree so that all calibration nodes have an age which is compatible with the calibrations set in the analysis.

Useful Links
Relevant References
- Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C.-H., Xie, D., Suchard, M. A., Rambaut, A., & Drummond, A. J. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Computational Biology, 10(4), e1003537. https://doi.org/10.1371/journal.pcbi.1003537
- Bouckaert, R., Vaughan, T. G., Barido-Sottani, J., Duchêne, S., Fourment, M., Gavryushkina, A., Heled, J., Jones, G., Kühnert, D., Maio, N. D., Matschiner, M., Mendes, F. K., Müller, N. F., Ogilvie, H. A., Plessis, L. du, Popinga, A., Rambaut, A., Rasmussen, D., Siveroni, I., … Drummond, A. J. (2019). BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLOS Computational Biology, 15(4).
- Drummond, A. J., & Bouckaert, R. R. (2014). Bayesian evolutionary analysis with BEAST 2. Cambridge University Press.
Citation
If you found Taming the BEAST helpful in designing your research, please cite the following paper:
Joëlle Barido-Sottani, Veronika Bošková, Louis du Plessis, Denise Kühnert, Carsten Magnus, Venelin Mitov, Nicola F. Müller, Jūlija Pečerska, David A. Rasmussen, Chi Zhang, Alexei J. Drummond, Tracy A. Heath, Oliver G. Pybus, Timothy G. Vaughan, Tanja Stadler (2018). Taming the BEAST – A community teaching material resource for BEAST 2. Systematic Biology, 67(1), 170–-174. doi: 10.1093/sysbio/syx060