

Background
Phylogenetic trees are often used to describe the history of genetic sequences. There are however several processes that recombine genetic material. Different parts of genome can then code for different histories and the shared history of the full genome can not be represented anymore by a tree. Reassortment is such a process that can reshuffle segments when people are infected by multiple viruses.
The coalescent with reassortment models a joint coalescent and reassortment process (Müller et al., 2020). To do so, it models how network lineages coalesce and recombine backwards in time.
In order to perform inference under the coalescent with reassortment, CoalRe uses MCMC sampling of the reassortment network and the embedding of segment trees within those networks. The way CoalRe is implemented, allows following the usual setup for BEAST2 xml files in BEAUti. The difference in setting up CoalRe compare to other models is that the Coalescent with Reassortment has to be specified as a tree prior for all segments individually.
After running CoalRe, the post-processing works slightly different to other models. Since we have to analyse a network and not just a tree, CoalRe implements a BEAST2 app that summarizes networks and works similar to treeAnnotator.
Programs used in this Exercise
BEAST2 - Bayesian Evolutionary Analysis Sampling Trees 2
BEAST2 (http://www.beast2.org) is a free software package for Bayesian evolutionary analysis of molecular sequences using MCMC and strictly oriented toward inference using rooted, time-measured phylogenetic trees. This tutorial is written for BEAST v2.6.0 (Drummond & Bouckaert, 2014).
BEAUti2 - Bayesian Evolutionary Analysis Utility
BEAUti2 is a graphical user interface tool for generating BEAST2 XML configuration files.
Both BEAST2 and BEAUti2 are Java programs, which means that the exact same code runs on all platforms. For us it simply means that the interface will be the same on all platforms. The screenshots used in this tutorial are taken on a Mac OS X computer; however, both programs will have the same layout and functionality on both Windows and Linux. BEAUti2 is provided as a part of the BEAST2 package so you do not need to install it separately.
IcyTree.org
IcyTree.org is a webbased tree viewer that, additional to trees, also allows to visualize networks in the extended newick format.
Practical: Setting up an coalescent with reassortment analysis
The coalescent with reassortment is an approach that allows inferring reassortment networks and rates of segmented viruses from the genetic sequences of individual segments.
In this tutorial, we will learn how to create an xml for a coalescent with reassortment analysis, how to then run this xml, as well as how to process and visualize the output of such an analysis.
The Data
The data consists of sequences from two segments (HA and NA) of the 2009 pandemic like Influenza A/H1N1 virus. HA (Hemagglutinin) and NA (Neuraminidase) are the two surface protein of influenza viruses. The genetic code for which sits on two different segments which can reassort. 2009 pandemic like Influenza A/H1N1, as the name suggests, caused the 2009 H1N1 pandemic. After, it became a seasonally circulating virus that has been circulating in the human population since.
Overall, the dataset we use in this tutorial consists of 25 HA and NA sequences of pandemic 2009 like human influenza A/H1N1 sampled between 2012 and 2017 downloaded from fludb.org. The sequences are already aligned and can be found in the data folder.
Download CoalRe
First, we have to download the CoalRe package, using the BEAUti package manager.
To do so, open BEAUTi and then go to File >> Manage Packages and download the CoalRe package .

After the package is installed, re-start BEAUti.
Load in the sequences
In order to load in the sequences into BEAUti, they can be dragged and dropped into the partitions window. Then a window will pop up, where we’ll have to specify that all the sequence files just loaded in are nucleotide sequences. This is so BEAUti knows which site models to suggest.
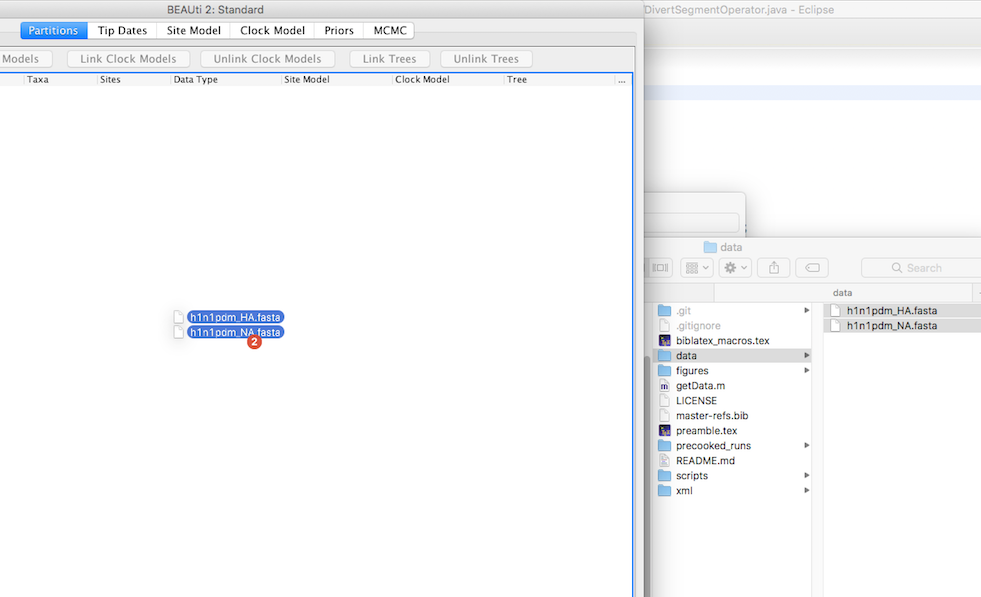
In order to account for rate variations across the different nucleotide sites, we next have to split both alignments into codon positions. To do so, select one of the segments and the press split and select the 1,2 + 3 option. This assigns the same relative evolutionary rate to the first 2 codon postitions and a different one for the third position. Changes in nucleotides on the third codon position are much less likely to result in changes in the amino acid composition of the corresponding virus. As such, the third position is more flexible and nucleotide changes are typically more likely to occur on these positions.
Next, repeat the same thing for the other segment.
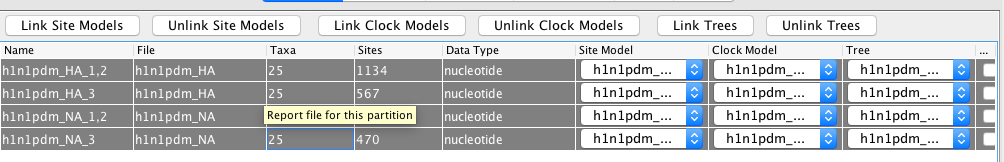
Linking the site models and clock models
Next, we’ll have to select all partitions, and then press Link Site Models, Link Clock Models.
Linking Clock models leads every partition to have the same evolutionary rate.
Linking the site model assigns every partition the same site model.
Assigning every partition the same site model is only done temporarily to speed up the setting up of the xml file and the site models will be unlinked later again.
Setting up the sampling times
Next, we’ll have to set up the sampling times.
To do so, go to Tip Dates and select Use tip dates
Specify the dates format to be yyyy-M-dd and press the Auto-configure button.
In the window that should pop up, select split on character

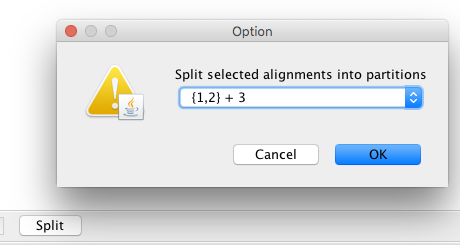
In order to set the tip dates for both segments, select both partitions an press OK to clone the tip dates
Setting up the site model
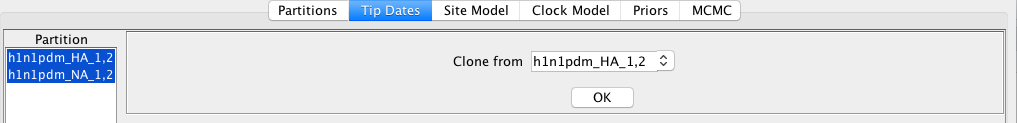
As a site model, we will use an $HKY+\Gamma_4$ model.
To do so, first set the site model from JC69 to HKY, which allows transition and transversion rates to differ.
Next, set the Gamma Category Count to 4 and make sure to click estimate for the Substitution Rate.
Setting the Gamma Category Count to 4 uses a discretized version of a gamma distribution to model different rate categories across sites.
4 or 5 different categories has been shown to be a good compromise between computational efficiency and still being able to approximate the gamma distribution well enough in practice.
Clicking estimate for the Substitution Rate allows each segment, as well as the first two and the third codon position to have a different relative rate of evolution.
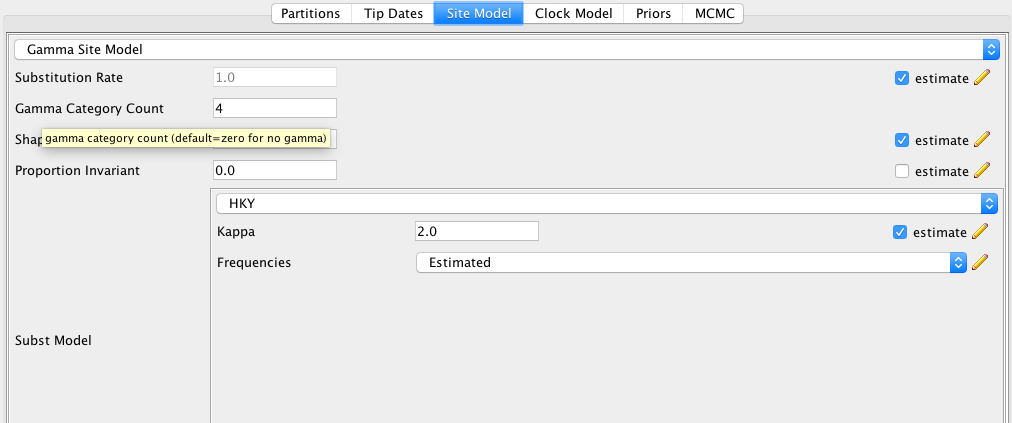
We next have to go back to the Partitions ta, select all Partitions and then press Unlink Site Models.
As mentioned above, the linking of the site models was only done to speed up setting up the xml.
After we unlinked the site models, each partition will have a site model that is setup the same way, but that has parameters that can be independently estimated.
As mentioned above, linking the site models initially was only done to speed up setting up the xml.
Setting up the Priors
We can leave the clock model as is, which means that we use a Strict Clock Model (default) and can directly go the the Priors tab.
The first and most important thing we have to do here, is to to change the Yule Model to Coalescent With Reassortment Constant Population.
This has to be done for both (!) segment trees, meaning there overall should be two Coalescent With Reassortment Constant Population.
This ensures that both segment trees are linked to a network in which they are embedded and that that network is linked to a coalescent with reassortment network prior.

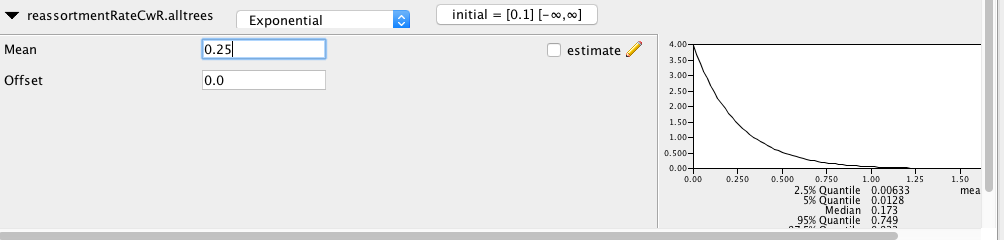
We next have to set the prior distribution on the parameters. The prior distribution on the effective population size can be left as is, but we have to change the prior on the reassortment rate. Set the prior distribution on the reassortment rate to be an exponential distribution with mean 0.25. This means that we assume a priori that on average there is one reassortment event per lineage occurring every 4 years. In previous analysis using similar datasets, this range of parameters has been shown to be what we can expect for influenza viruses (Müller et al., 2020).
Setting up the Chain Length
Next, we can go to the MCMC panel to set the chain length. For this analysis with just a few sequences, a chain length of 5 million should be enough.
This was the last step of setting up the xml and we can now save it by going to File > Save as
Run the xml
Next, open BEAST and run the xml.
This should take somewhere in the order of 15 to 20 minutes.
Alternative, the folder precooked_runs contains the log files of the run.
Inspect the run in Tracer
Next, we have to check whether everything converged.
To do so, we can open the program Tracer and load the *.log file.
All ESS values should optimally be above 200.
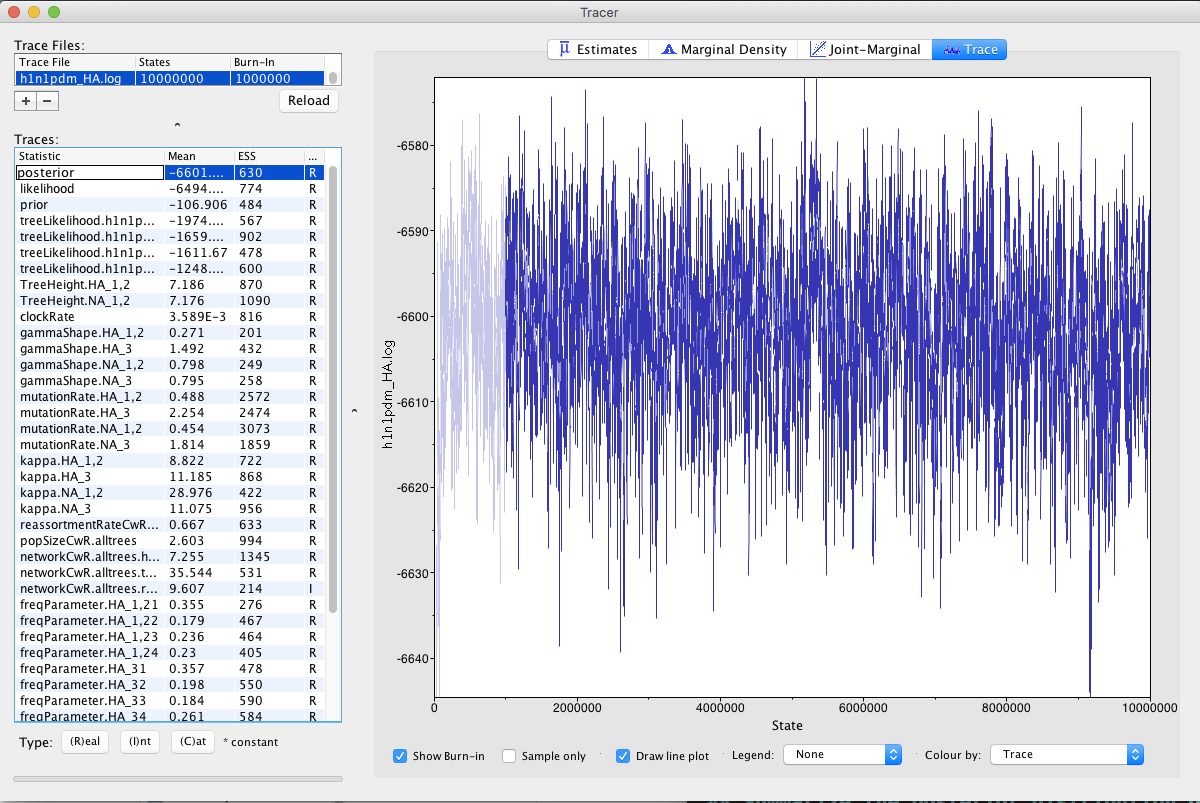
Next, we can check what rates were inferred.
The clockRate denotes the average rate of evolution across all segments.
The reassortmentRateCwR.alltrees denotes the rate of reassortment (from present to past) per lineage and year.
Summarize the posterior distribution of networks
Next, we can summarize the distribution of networks by maximizing the clade credibilities.
To do so, open BEAUti and select File > Launch Apps.
Then, select Reassortment Network Annotator.
Next, choose the networks.trees file as input for the Reassortment Network log file and choose the file where the mcc network should be saved to and press analyse.
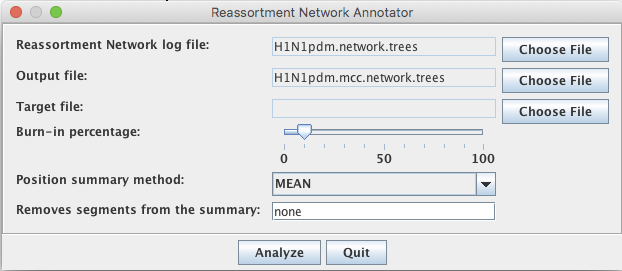
The option ‘Removes segments from the summary’ is not relevant for datasets with only 2 segments. When there are more segments, however, it can e.g. be interesting to look at the reassortment network of pairs of segments. This option allows to remove segments from an analysis. Keep in mind though that the numbering of segments is not neccessarily the biological one, but the alphabetical one (for implementation reasons).
Visualize the network using icytree.org
Next, open your browser and go to the webpage icytree.org(Vaughan, 2017) The resulting mcc network file can now be drag and dropped into icytree to visualize the network. Icytree plots the network as a base tree that is connected by dotted branches. This implies that at a reassortment event, there is a difference between the two parent branches. This is, however, not the case in the coalescent with reassortment model, but for simplicity is plotted like this. The “main” branch here is the always the parent branch that carries more segments. If both branches carry the same amount of segments, the branch that is closer the next event is chosen as the main branch.
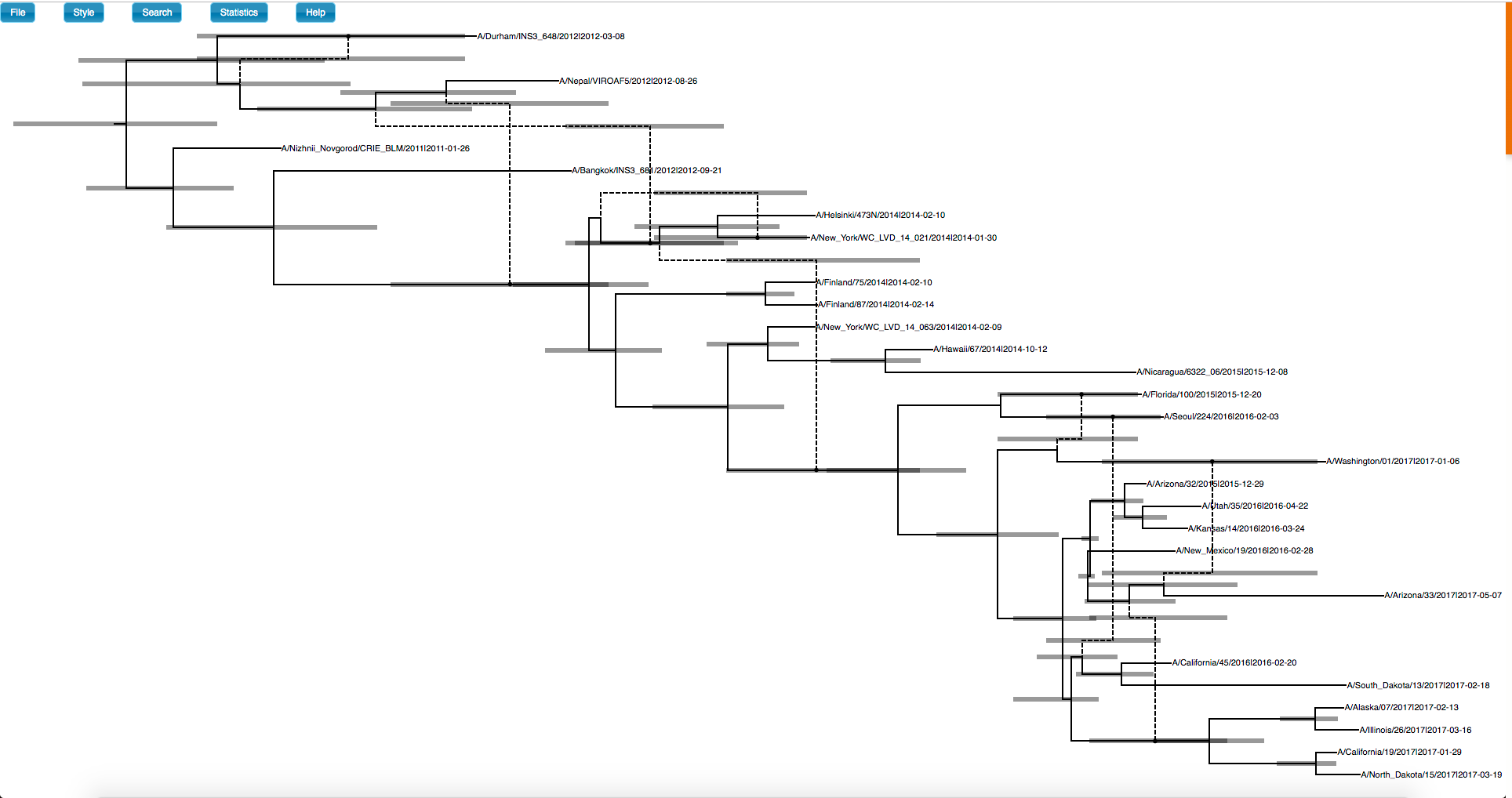
The 95 $\%$ highest posterior density intervals for node heights can be plotted by going to Style > Node height error bars.
The posterior support for each node can be shown by going to Style > Internal node text.
Useful Links
- BEAST 2 website and documentation: http://www.beast2.org/
- Join the BEAST user discussion: http://groups.google.com/group/beast-users
Relevant References
- Müller, N. F., Stolz, U., Dudas, G., Stadler, T., & Vaughan, T. G. (2020). Bayesian inference of reassortment networks reveals fitness benefits of reassortment in human influenza viruses. Proceedings of the National Academy of Sciences.
- Drummond, A. J., & Bouckaert, R. R. (2014). Bayesian evolutionary analysis with BEAST 2. Cambridge University Press.
- Vaughan, T. G. (2017). IcyTree: Rapid browser-based visualization for phylogenetic trees and networks. Bioinformatics. https://doi.org/10.1093/bioinformatics/btx155
Citation
If you found Taming the BEAST helpful in designing your research, please cite the following paper:
Joëlle Barido-Sottani, Veronika Bošková, Louis du Plessis, Denise Kühnert, Carsten Magnus, Venelin Mitov, Nicola F. Müller, Jūlija Pečerska, David A. Rasmussen, Chi Zhang, Alexei J. Drummond, Tracy A. Heath, Oliver G. Pybus, Timothy G. Vaughan, Tanja Stadler (2018). Taming the BEAST – A community teaching material resource for BEAST 2. Systematic Biology, 67(1), 170–-174. doi: 10.1093/sysbio/syx060