

Background
Bacter is a BEAST 2 package dedicated to performing inference of bacterial sequence data. In general, it allows for joint inference of the Ancestral Recombination Graph (ARG), recombination rates, tract lengths, substitution rate and population dynamics. Exactly what may be inferred from a given data set depends heavily on the size of the data set, and the rates of recombination and mutation for the population under study, and the sampling procedure.
This tutorial will gently walk you through the process of using Bacter to the ARG and associated parameters using an example multi-locus data set.
Installation
Software requirements
In order to use Bacter and visualize its analysis results the following software must be installed:
- BEAST 2 version 2.5 or later. http://www.beast2.org/
- Tracer version 1.7 or later. https://github.com/beast-dev/tracer/releases/latest
- A recent version of Mozilla Firefox or Google Chrome.
Bacter Package Installation
Bacter is easily installed via the BEAUti package manager. To do this, run BEAUti and select “Manage Packages” from the File menu:
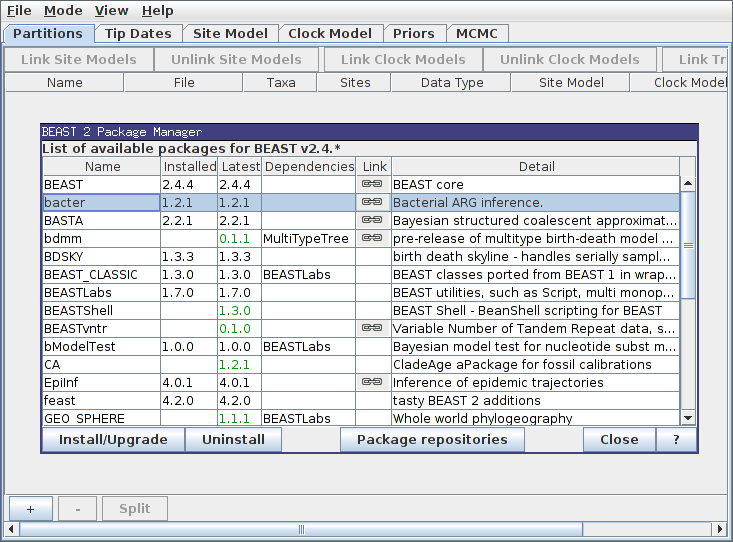
Then, ensure the “bacter” package is highlighted before pressing the “Install/Upgrade” button:
That’s it! Bacter is now installed. It is a good idea to restart BEAUti at this point.
Setting up the analysis
Choosing the Bacter BEAUti template
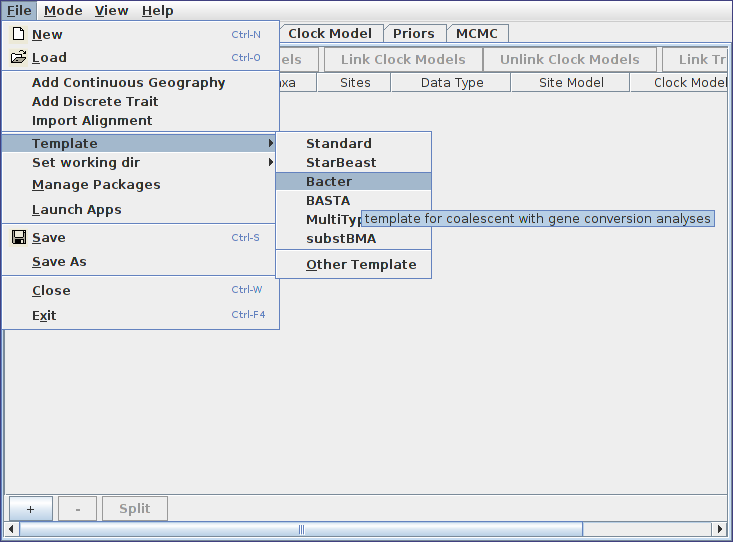
Open the BEAUti program. Before doing anything else we must switch to the Bacter template. To do this, open the file menu and from the Template submenu select “Bacter”.
Loading sequence alignments
In this tutorial we will be analyzing a subset of the E. coli data presented in Vaughan et al., Genetics, 2017. The full data set consists of 53 individual gene alignments, each containing 23 sequences. In order to keep our tutorial analysis as manageable as possible, we will analyze only 12 of these genes.
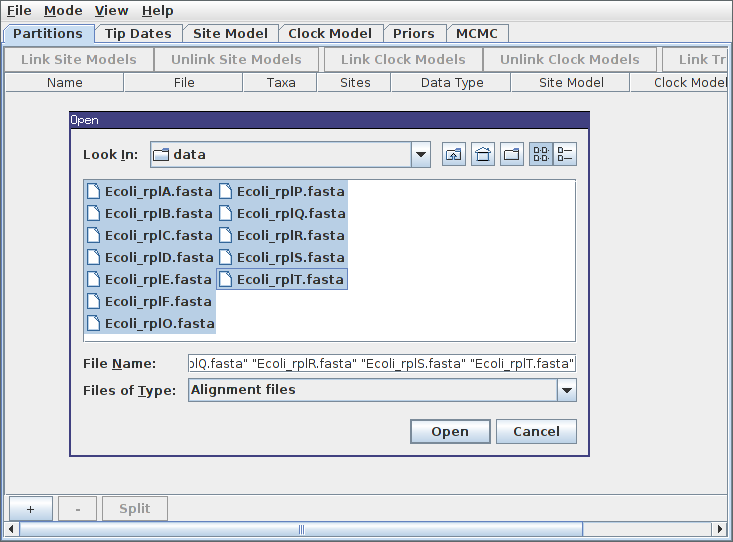
To load the alignments, select File->Import Alignment and navigate to the directory containing the tutorial data. This directory contains 12 FASTA files, each containing the alignment for the named gene. Since the directory contains only these files and nothing else, we can select them all simply using Ctrl+A (or Command+A on a Mac).
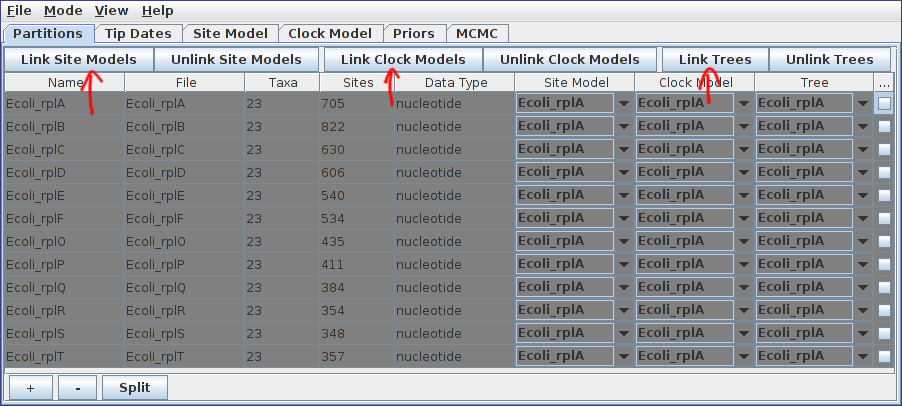
After pressing Open, the alignments should be visible as 12 new records in the table. By default, each locus is assumed to have its own distinct site, clock and “tree” (really ARG here) models. Since our ARGs potentially span multiple loci, we should definitely cause the loci to share a tree model. This is done by selecting both rows in the table by again holding down Ctrl (or Command) and clicking each in turn. Alternatively, you can just click on one row and then press Ctrl+A, or Command+A on a Mac. (This is easier than clicking each row if your data consists of many loci.) Once they’re selected, press the “Link Trees” button on the right-hand side just above the table. In our case, we will use shared site and clock models too, so click the “Link Site Models” and “Link Clock Models” buttons as well.
Defining the model
We will now configure the model under which the inference will be conducted.
The data that we’ve loaded was sampled contemporaneously. We can therefore ignore the Tip Dates panel. When analyzing data where the samples were collected at very different times you’ll want to include those times in the analysis by modifying the contents of that panel.
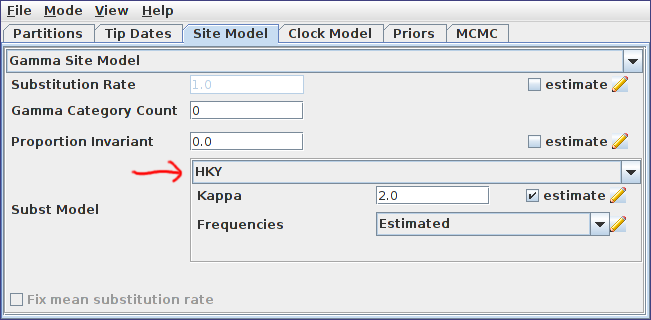
Open the Site Model panel and set the substitution to HKY. This model is far superior to the default Jukes-Cantor model as it allows distinct transition/transversion rates and non-equal equilibrium base frequencies. Leave the default initial value for kappa and the base frequencies as “estimated”. The site model panel should now look similar to the following:
Again, because the data was sampled contemporaneously and we have no relevant calibration information, we will ignore the Clock Model panel. By doing this, we are implicitly deciding that time will be expressed in expected number of substitutions per site.
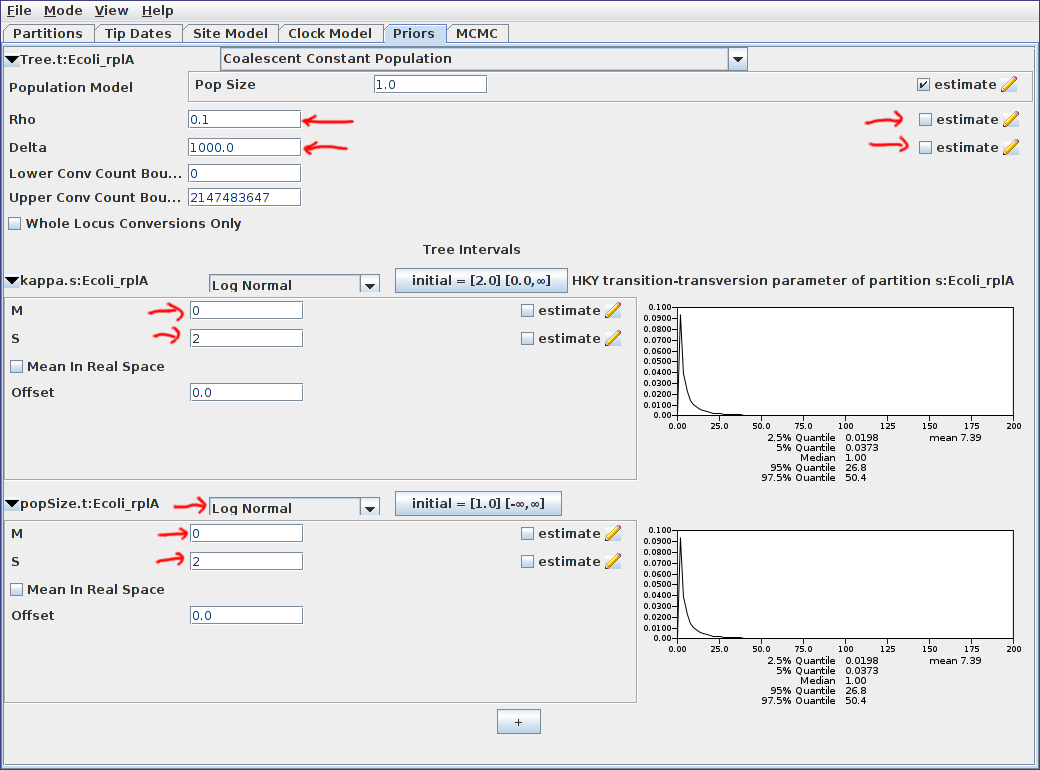
Now switch to the Priors panel. See that “Coalescent with constant population” is selected as the default tree prior. Expand this tree prior by clicking on the arrow to the left of “Tree.t”.
Uncheck the “estimate” checkboxes to the right of the recombination rate (Rho) and tract length (Delta) parameters and enter the values 0.1 and 1000.0 respectively. Estimating these parameters can actually be quite difficult, so fixing them to known values is a good idea if these are available. In this particular case, while 1000 is a reasonable value for the expected conversion tract length of E. Coli, fixing the recombination rate to 0.1 is suspect. We do this solely to reduce the computation time required for this analysis.
Select a log normal prior for the population final size parameter, with parameters M=0 and S=2.
The priors panel should now look similar to the following:
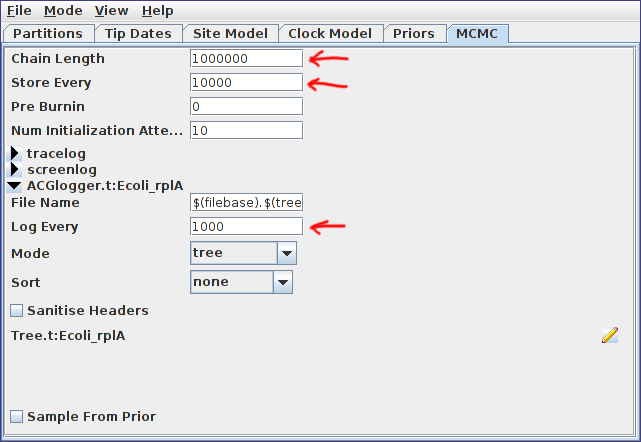
Finally, switch to the MCMC panel and change the chain length to 1000000 (i.e. 10^6). This is far too short for a production run, but means that the example analysis will complete relatively quickly. Also, because our run is so short, we’ll sample the chain of ARGs more frequently. To do this, expand the ACGLogger.t:simulated_data_A section by clicking on the arrow to the left of this line, then set the “Log Every” field to 1000. The MCMC panel should now look similar to the following:
Once your analysis is set up, select File->Save, navigate to the directory you wish the analysis XML to be written to, give it a sensible file name (for example bacter_tutorial.xml), and press the Save button to produce the BEAST input XML.
Note: If you experience difficulties using BEAUti to generate the BEAST XML file but would like to continue immediately with the analysis, download and use this ready-made XML file;
Running the analysis
Run the analysis just as you would any other BEAST 2 analysis. That is,
- Start BEAST 2.
- Select the XML you produced in the previous section from the file selection dialog box.
Once BEAST is running, you should see output periodically printed to standard out (if you’re running BEAST from a terminal emulator) or the output window. The analysis we’ve set up should take 15 minutes to complete on a modern computer.
If you run out of time to complete the analysis, simply download the pre-cooked log and tree files and continue the tutorial using them.
Analyzing the results
During the analysis results are written to several files which can usually located in the same directory as the directory containing the input XML. These are:
- The log file, which ends in the extension .log and contains sampled parameter values,
- The tree file, which ends in the extension .trees and contains sampled ARGs.
Parameter posteriors
To examine the sampled parameter marginal posteriors and, more generally, to assess the health of our analysis results, open Tracer and load the log file. (To do this, select “Import trace file” from the File menu or click the “+” button below and to the left of the table in the top left corner of the Tracer window.) Shown below is the marginal posterior for the number of conversions ancestral to our data.
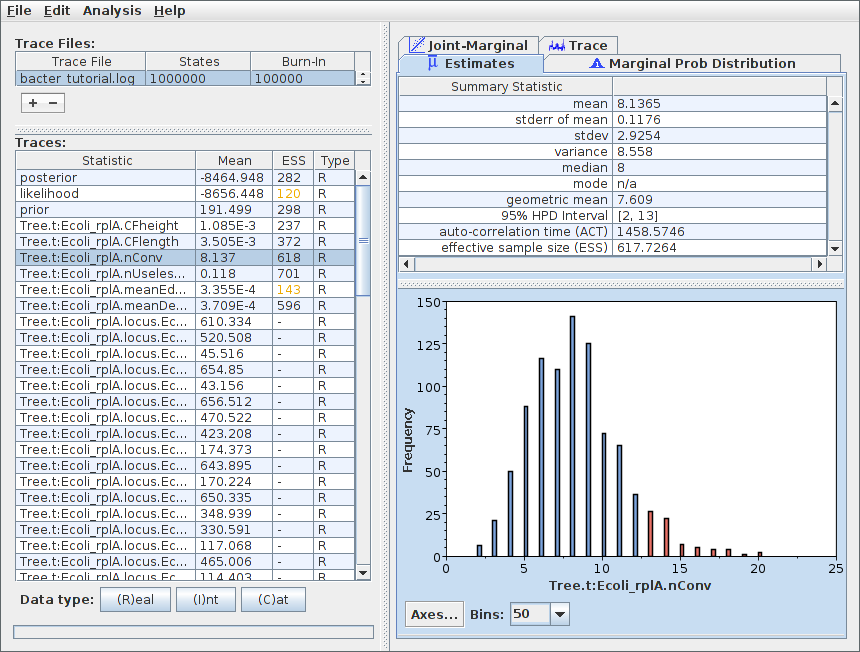
Note that the ESS for many parameters may still be extremely small after 10^6 iterations. This indicates that, as anticipated, the chain should be run for a lot longer before the results are considered trustworthy. We can easily continue/resume the chain just as for any other BEAST 2 analysis, and you may wish to try this yourself. However, in the interests of keeping the length of time needed to complete this tutorial from becoming too long, we will now proceed to further analyse the results we already have.
Viewing sampled ARGs
The ARGs sampled during a Bacter analysis can be viewed using the browser-based IcyTree phylogenetic tree and network visualizer. Beware that the viewer requires an up-to-date version of Firefox or Chrome to function correctly.
To use the viewer, simply open the IcyTree web page in a browser window, select File->“Load from file”, then choose the tree file using the file chooser. Alternatively, you can simply drag the tree file onto the IcyTree window.
Once loaded, the first ARG in the tree file is displayed. Use the comma and period (, and .) keys to step through the file one ARG at a time or the < and > keys to step in increments of 10%. Navigation can also be performed by clicking on the buttons in the lower-left corner of the window with your mouse. Further information about using IcyTree can be found by selecting items listed under the Help menu. To generate the image below, edges were coloured by locus (Style->“Colour edges by”), the colouring legend and the time axis were switched on (Style->“Display legend” and Style->“Display axis”).
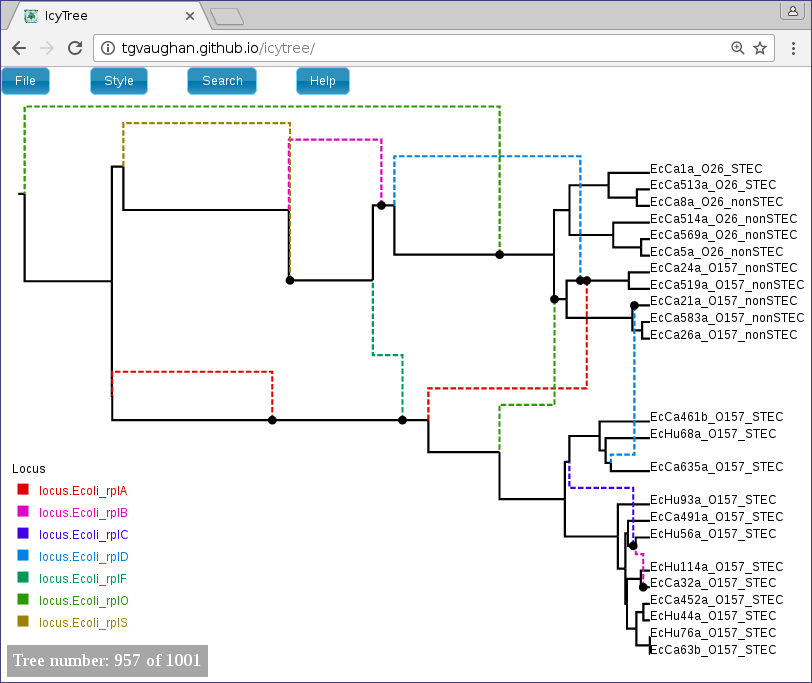
ARGs are displayed in IcyTree in a particular way. The solid lines depict lineages belonging to the clonal frame, while dashed edges representing the topology changes imposed on the clonal frame by conversions. Additional information concerning a specific edge can be viewed by hovering the mouse cursor over that edge.
It is important to remember that ARGs at the start of the file (particularly the first) will likely be very different to the true ARG, as this portion of the file represents ARGs sampled before convergence of the MCMC to the true posterior. Later trees should represent individual samples drawn from the posterior.
Creating a summary ARG
Individual ARGs sampled from the posterior are poor representations of the inference result at best, and at worst they may be completely misleading. This is because they contain no indication of the uncertainty inherent in what the sequence data tells us of the events they describe. Thus, while a single sampled ARG may contain features that are well-supported by the data, the same ARG will likely contain many features that have little or no support at all.
What is needed is some kind of picture of the posterior distribution over ARG space instead of a single point estimate. Unfortunately, the optimal route to producing such a summary is currently an open research question. However, Bacter provides an implementation of an algorithm for constructing a qualitative summary which is similar in spirit to the algorithms which BEAST and other Bayesian phylogenetic packages use to summarize distributions over tree space. (Refer to the paper for details.)
To produce a summary ARG, open the “AppLauncher” program that is distributed with BEAST 2. (You can also select “Launch Apps” from the File menu in BEAUti.)
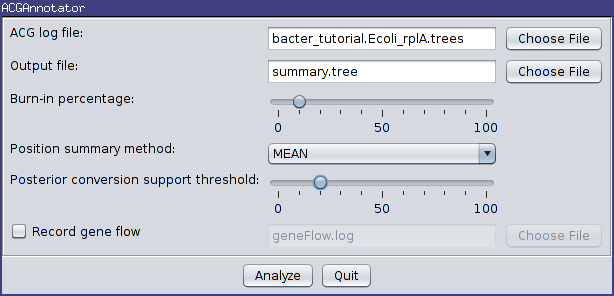
Ensure the ACGAnnotator icon is highlighted, then press the Launch button. This will open a dialog box from which you can select the ACG (tree) log file and the name and location of the file to which the summary is written. In addition, you can adjust:
- The burn-in percentage: the percentage of the tree log file which will be ignored to account for the samples that were taken before the MCMC reached convergence.
- The position summary method: this affects the way that the internal node ages of the summary tree are chosen. The default is to use the mean of the node ages present in the sampled ARGs.
- The posterior conversion support threshold: this is the percentage of sampled ARGs which must contain a given conversion for it to be included in the summary tree. (This is conceptually similar to the threshold for clade inclusion in a majority-rule consensus tree.)
For this tutorial, ensure the ACG (tree) log file generated by the above analysis is selected and choose summary.tree as the output file name. Leave the burn-in fraction at 10% and keep MEAN as the position summary method, but lower the conversion support threshold to 20%. This means that conversions will only have to appear in 20% of the sampled ARGs to be included in the summary. The dialog box should now look like the following image:
Pressing the “Analyze” button will bring up an additional window which will report on the progress of creating the summary tree. As there are only a few hundred ARGs present in our log file, this process should only take a few seconds. Once it is complete, press the Close button. You can also exit the AppLauncher.
Loading the file summary.tree in IcyTree should produce something similar to the following figure. (Edges have been coloured by “locus”, and labelled with their posterior support. Error bars indicating the 95% HPD intervals for the age of each ancestral event have also been included.)
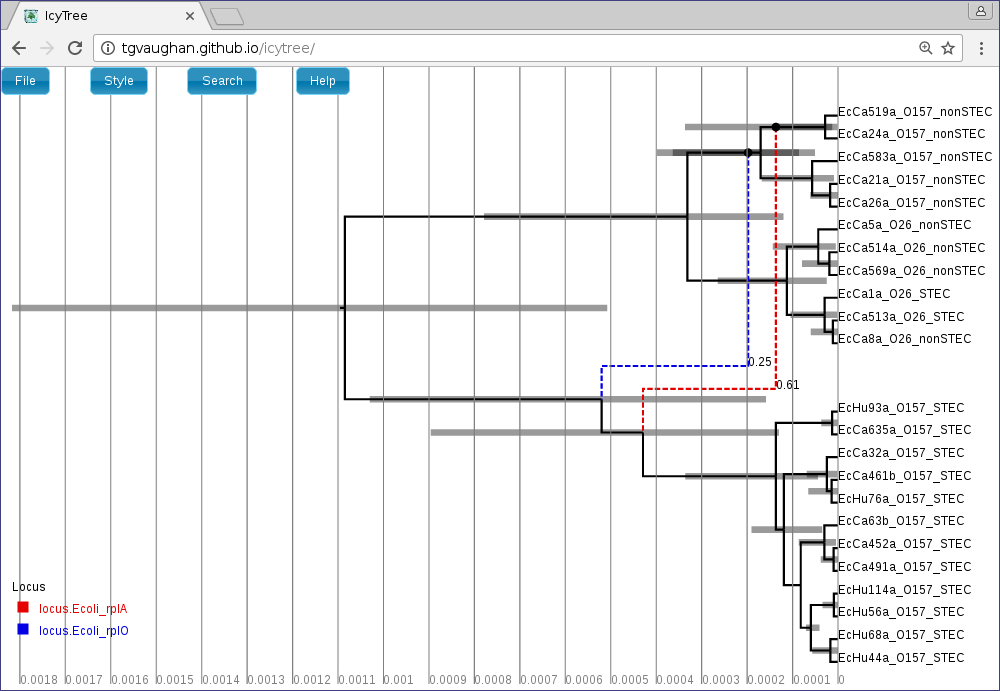
This summary suggests that our E. coli dataset only has evidence for one or two conversions: one on the rplA gene with ~60% support and one on the rplO gene with much lower support. Note however, that this summary does not at all rule out the possibility of many other conversion events ancestral to the data: it simply indicates that only these conversions occur in roughly the same place in at least 20% of the posterior samples.
This distinction is backed up by the marginal posterior distribution for the number of conversion events in the graph shown in the tracer figure above, which has a median of 8 events - still very small, but four times the number appearing in the summary. The discrepancy is likely made up of conversions that produced very little or no signal in the data.
Wrapping up
This completes our introductory Bacter tutorial. There are other features that are not covered here, such as the ability to perform inference under different parametric and non-parametric models of population dynamics. Tutorials explaining these features will appear in the near future on the Bacter web page.
Useful Links
- Bacter website and documentation: https://tgvaughan.github.io/bacter
- BEAST 2 website and documentation: http://www.beast2.org/
- Join the BEAST user discussion: http://groups.google.com/group/beast-users
Citation
If you found Taming the BEAST helpful in designing your research, please cite the following paper:
Joëlle Barido-Sottani, Veronika Bošková, Louis du Plessis, Denise Kühnert, Carsten Magnus, Venelin Mitov, Nicola F. Müller, Jūlija Pečerska, David A. Rasmussen, Chi Zhang, Alexei J. Drummond, Tracy A. Heath, Oliver G. Pybus, Timothy G. Vaughan, Tanja Stadler (2018). Taming the BEAST – A community teaching material resource for BEAST 2. Systematic Biology, 67(1), 170–-174. doi: 10.1093/sysbio/syx060